 Prof. Univ. Dr. Petre Cornel Brătilă
Prof. Univ. Dr. Petre Cornel BrătilăAuthors
Introducere
Sindromul Mayer-Rokitansky-Kuster-Hauser (MRKH) este o afecțiune congenitală rară, cu o incidență de 1 la 5000 de nou-născuți de sex feminine (1,2) caracterizat prin aplazia congenitală a uterului și cele 2/3 superioare ale vaginului la femei cu caractere sexuale secundare normal reprezentate și un cariotip normal 46, XX(3). Pacientele pot prezenta fie aplazie utero-vaginală izolată (tipul I), fie pot asocia diferite malformații extragenitale (tipul II) (4). Principalul simptom care trădează MRKH este amenoreea primară la o pacientă cu organe genitale externe și caractere sexuale secundare normal reprezentate (5). Impactul pe care îl are această afecțiune asupra pacientelor este unul semnificativ, atât fizic cât și psihologic, cauzat în principal de afectarea fertilității (6,7).
Acest articol are ca scop prezentarea unui caz rar de sindrom MRKH diagnosticat într-un mod necaracteristic, având ca motiv principal al prezentării în clinică prolapsul de boltă vaginală.

Prezentare caz clinic
Pacienta se prezintă pentru prima dată în clinica noastră la vârsta de 23 de ani, cu senzație de presiune la nivelul introitului vaginal, senzație de protuzie a pereților vaginali, dispareunie și durere sacrală la ortostatism prelungit. Din anamneză nu reținem antecedente heredo-colaterale semnificative.
Istoricul medical al pacientei este complicat. La vârsta de 10 ani a fost diagnosticată cu tiroidită cronică autoimună cu hipotiroidism. La 14 ani se stabilește diagnosticul de amenoree primară, fără istoric familial asemănător, fiind infirmate afecțiuni materne pe timpul sarcinii sau expunerea intrauterină la tratament cu efect teratogen.
La vârsta de 22 de ani, pacienta dezvoltă prolaps de boltă vaginală în urma primului contact sexual. Se prezintă la consult și este diagnosticată cu sindrom MRKH prin examen IRM abdomino-pelvin.
În martie 2017 se practică în altă clinică colposuspensia sacrospinoasă, pe cale laparoscopică cu fire, cu evoluție nefavorabilă și recidiva prolapsului. Se reintervine chirurgical și se practică colposacrosuspensie laparoscopică, urmată în evoluție de un episod de sepsis cu etiologie neelucidată. La o lună de la intervenție, se intervine chirurgical pentru chist pilonidal abcedat. Două luni mai târziu, investigațiile imagistice relevă UHN dreapta grad II-III, sacrodiscită, spină bifidă incompletă. Se montează sondă ureterală dreapta JJ.
La internarea în clinica noastră, examenul clinic general relevă pacienta normoponderală, cu caractere sexuale secundare stadiul 5 Tanner, corespunzătoare vârstei. Sistemul musculo-scheletal este integru, fără modificări.
Examenul clinic local pune în evidență prezența unui vagin scurt, de 4 cm (prezintă doar vaginul inferior cu pliuri parțial păstrate), prolabat la nivelul vulvei, care se termină în fund de sac.

Imagine 1. Prolaps de boltă vaginală
Se efectuează RMN abdomino-pelvin nativ și cu CIV ce arată acumulare neomogenă cu aspect fibrotic inflamator situată presacrat (anterior de spațiul intervertebral L5-S1 paramedian drept) ce interesează în marginea ei și ureterul drept pe o lungime de cca 3 cm, prezintă modificări aderențiale față de ovarul drept și o comunicare cu o colecție situată posterior de ligamentul prevertebral la nivelul spațiului discal L5-S1, spondilodiscită L5-S1, uter cu dimensiuni mult reduse, ovare si trompe uterine prezente.

Imagine 4. IRM pelvin
În urma corelării elementelor clinco-imagistice se stabilește diagnosticul actual de prolaps boltă vaginală recidivat, stenoză ureterală dreaptă protezată cu stent pentru UHN gr. II-III, sacrodiscită, malformație complexă utero-cervico-vaginală U5bC4V4 (Sindrom MRKH).
Se intervine chirurgical și se practică ureteroliză dreaptă, excizia meșei de polipropilena pe cale abdominală (pentru rezolvarea focarului infecțios și consecințelor sale) și colposuspensia la ligamentele sacrospinoase pe cale vaginală, pentru rezolvarea prolapsului genital.
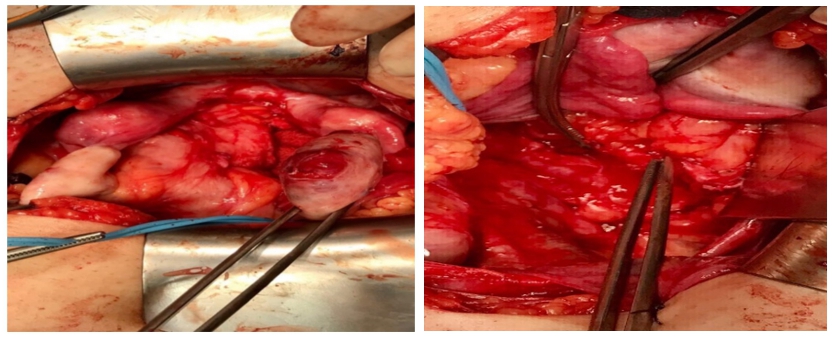
Imaginile 5 și 6: Aspect intraoperator
Sonda ureterală se extrage cistoscopic în octombrie 2017. Un an și jumatate mai târziu, debutează o nouă simptomatologie, manifestată prin stare subfebrilă, urmată 2 săptămâni mai târziu de apariția durerii în regiunea pelviană și de secreții vaginale cu caracter purulent. Starea pacientei s-a agravat cu 48 de ore înainte de prezentarea în clinica noastră prin alterarea marcată a stării generale, febră (40oC), apariția de secreție purulentă fetidă în vagin, semne inflamatorii vulvare drepte, oligurie, fatigabilitate și inapetență.
La examenul ginecologic se constată vulva cu edem important al labiei drepte, cu semne celsiene prezente, bont vaginal normal situat, cu 2 orificii fistuloase la nivelul peretelui vaginal drept și la nivelul boltei vaginale, prin care se exteriorizează puroi galben-cenușiu, fetid. Biologic se constată leucocitoză 31300/mm3. Corelarea cu examinarea IRM pune diagnosticul de abces paravaginal fistulizat, cu extensie la nivelul foselor obturatorii bilateral, intervezicomezorectal și vulvar. Evoluția sub triplă terapie antibiotică sistemică, reechilibrare hidroelectrolitică și lavaj local cu soluții antibiotice este favorabilă, cu remiterea totală a simptomatologiei.

În noiembrie 2019, pacienta se prezintă cu simptome identice episodului precedent. Examenul ginecologic relevă vulva de aspect normal, bont vaginal bombat, cu prezența a două orificii fistuloase la nivelul peretelui vaginal drept și la nivelul boltei vaginale, prin care se exteriorizează puroi. Biologic se constată leucocitoză 29200/mm3 . Corelarea cu examinarea IRM pune, pentru a doua oară, diagnosticul de abces paravaginal fistulizat, cu extensie la nivelul foselor obturatorii bilateral, intervezicomezorectal și vulvar. Se decide tratament conservator prin triplă terapie antibiotică sistemică, reechilibrare hidroelectrolitică și lavaj local cu solutii antibiotice, cu evoluție ulterioară rapid favorabilă și remiterea totală a simptomatologiei.
În februarie 2021, pacienta se prezintă pentru secreții vaginale purulente. Examenul ginecologic relevă vulva de aspect normal, bont vaginal normal situat, cu orificiu fistulos în comisura dreaptă, prin care se exteriorizează puroi gălbui și meșa de polipropilenă. După obținerea consimțământului informat se practică excizia meșei de colposuspensie, cu evoluție favorabilă până în prezent.
Discuții
Din perspectivă embriologică, uterul, colul uterin și cele două treimi superioare ale vaginului se formează din ductele paramezonefrice Mülleriene, în timp ce treimea inferioară a vaginului se dezvoltă din sinusul urogenital, în jurul săptămânii 5-6 de gestație (8,9). Anumite anomalii genetice pot afecta dezvoltarea embrionară, provocând agenezia sau aplazia completă a ductelor paramezonefrice, ceea ce duce la fenotipul sindromului MRKH (6).
Etiologia sindromului a fost o lungă perioadă de timp incertă. Inițial, a fost considerată o afecțiune sporadică, fiind luați în considerare factori de mediu non-genetici (10), diabetul gestational (11) sau medicamente cu efect teratogen precum Talidomida sau Dietilsilbestrolul (12,13), teorii infirmate ulterior prin analiza retrospectivă a istoricului sarcinilor (14,15,16). Rudele de gradul I ale pacienților cu sindrom MRKH au un risc de 1-5% de dezvoltare a unor anomalii congenitale uterine (17,18) sau renale (15,19), cu o limitare a transmiterii cu caracter autozomal dominant cu penetranță incompletă la sexul feminin, sugerând că defectul genetic este transmis pe linie paternă (20,21).
Caracteristicile principale ale sindromului MRKH sunt absența congenitală a uterului și a 2/3 superioare ale vaginului la paciente cu un cariotip normal (46, XX) (22) și funcție ovariană normală (4,23) fără asocierea hiperandrogenismului(24), toate aceste caracteristici fiind valabile și în cazul pacientei noastre: organe genitale externe normale și caracterele secundare sexuale normal reprezentate, sâni și păr pubian (stadiul 5 pe scala Tanner), vagin închis în fund de sac, cu o lungime de 2-7 cm.
Din punct de vedere anatomic, tipul I este caracterizat prin aplazia uterului și prezența a două relicvate a cornurilor uterine, conectate printr-un fald peritoneal, cu ovare și salpinge de aspect normal. Aspectul intraoperator, corelat cu datele imagistice și clinice, a fost sugestiv în cazul nostru pentru sindrom MRKH tip I. Tipul II este caracterizat de hipoplazie simetrică sau asimetrică a uterului, asociat cu aplazia unuia dintre cornurile uterine sau asimetrie de dimensiune dintre cele doua cornuri și malformatii ale salpingelor (3,25). Prezența relicvatelor uterine poate fi asociată cu manifestări catameniale, pelvialgii, hematometrie, criptomenoree (26), cauzate de activitatea endometrială (27,28), sau leziuni specifice endometriozei (29,30). Tipul II asociază cel mai frecvent malformații renale, scheletale sau ale urechii și rareori cardiac (31,32,33).
IRM pelvin este standardul de aur pentru diagnosticarea MRKH, evidențiind agenezia totală a structurilor Mülleriene, sau prezența relicvatelor și a endometrului de la nivelul acestora, prezența ovarelor, precum și alte eventuale malformații associate (34). În cazul nostru, examenul IRM a fost efectuat abia la 22 de ani, deși pacienta prezenta amenoree primară neinvestigată până la această vârstă.

Diagnosticul diferențial se face între afecțiuni ce cauzează amenoree primară la paciente cu caractere sexuale secundare normal reprezentate și include atrezia vaginală izolată, agenezia uterului și a vaginului și sindromul de rezistență la hormonii androgeni (35). Septul vaginal transvers sau himenul imperforat, care pot mima aspectul sindromului MRKH, prezintă uter și col indemne, ce pot fi palpabile transrectal sau pot fi vizualizate ecografic (3).
În prezent, malformațiile genitale se clasifică conform CONUTA-ESHRE.
Malformația prezentă a fost încadrată conform acestei clasificari după următoarele criterii:
- U5b- uter displazic (clasa 5) cu rudiment uterin bilateral fără cavitate (sub-clasa b)
- C4- aplazie cervicală
- V4- aplazie vaginală
Particularitatea acestui caz constă în apariția prolapsului de boltă vaginală ca prim simptom ce a determinat prezentarea la medic.
Agenezia uterină, a colului uterin și a celor 2/3 superioare ale vaginului implică absența nivelelor I și II de susținere (36,37). Nivelul I implică suspensia pereților vaginali laterali din porțiunea cefalică la pereții pelvini laterali prin intermediul fibrelor lungi de țesut conjunctiv ale paracolposului. În regiunea medie, fibrele paracolposului se scurtează, dar își păstrează funcția (38).
Pacienta a dezvoltat prolaps de boltă vaginală din cauza absenței nivelelor I și II de susținere a vaginului, în asociere cu factorii favorizanți reprezentați de efortul fizic intens și de contactul sexual. Lipsa structurilor de susținere și persistența factorilor de risc au dus la apariția complicațiilor in zona de ancorare sacrată L5-S1, fapt pentru care a fost îndepărtată meșa de polipropilenă și ancorarea bontului a fost făcută la ligamentele sacrospinoase.
Concluzie
Tratamentul chirurgical al prolapsului genital în cazul pacientelor cu Sindrom Rokitanski (malformații complexe utero-cervico-vaginale) este dificil, fiind însoțit de complicații a căror rezolvare pune probleme deosebite.

Bibliografie
- Ray U, Adhikari S, Dhital R, et al. Mayer-Rokitansky-Kuster-Hauser Syndrome: A rare case report from Nepal. Annals of Medicine & Surgery. 2022;82. doi:10.1016/j.amsu.2022.104725
- Herlin M, Bjørn AMB, Rasmussen M, Trolle B, Petersen MB. Prevalence and patient characteristics of Mayer–Rokitansky–Küster–Hauser syndrome: a nationwide registry-based study. Human Reproduction. 2016;31(10):2384-2390. doi:10.1093/humrep/dew220
- Morcel K, Camborieux L, Guerrier D. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Orphanet J Rare Dis. 2007;2(1):13. doi:10.1186/1750-1172-2-13
- Herlin MK, Petersen MB, Brännström M. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: a comprehensive update. Orphanet J Rare Dis. 2020;15(1):214. doi:10.1186/s13023-020-01491-9
- Nguyen BT, Dengler KL, Saunders RD. Mayer–Rokitansky–Kuster–Hauser Syndrome: A Unique Case Presentation. Mil Med. 2018;183(5-6):e266-e269. doi:10.1093/milmed/usx066
- Triantafyllidi VE, Mavrogianni D, Kalampalikis A, Litos M, Roidi S, Michala L. Identification of Genetic Causes in Mayer-Rokitansky-Küster-Hauser (MRKH) Syndrome: A Systematic Review of the Literature. Children. 2022;9(7):961. doi:10.3390/children9070961
- Deng S, He Y, Chen N, Zhu L. Spectrum of Type I and Type II Syndromes and Associated Malformations in Chinese Patients with Mayer-Rokitansky-Küster-Hauser Syndrome: A Retrospective Analysis of 274 Cases. J Pediatr Adolesc Gynecol. 2019;32(3):284-287. doi:10.1016/j.jpag.2018.07.007
- Kobayashi A, Behringer RR. Developmental genetics of the female reproductive tract in mammals. Nat Rev Genet. 2003;4(12):969-980. doi:10.1038/nrg1225
- Cunha GR, Robboy SJ, Kurita T, et al. Development of the human female reproductive tract. Differentiation. 2018;103:46-65. doi:10.1016/j.diff.2018.09.001
- Simpson JL. Genetics of the female reproductive ducts. Am J Med Genet. 2000;89(4):224-239. doi:10.1002/(SICI)1096-8628(19991229)89:4<224::AID-AJMG7>3.0.CO;2-C
- Martinez-Frias ML, Bermejo E, Rodriguez-Pinilla E, Prieto L, Frias JL. Epidemiological analysis of outcomes of pregnancy in gestational diabetic mothers. Am J Med Genet. 1998;78(2):140-145. doi:10.1002/(SICI)1096-8628(19980630)78:2<140::AID-AJMG8>3.0.CO;2-S
- HOFFMANN W. THALIDOMIDE AND FEMALE GENITAL MALFORMATIONS? The Lancet. 1976;308(7989):794. doi:10.1016/S0140-6736(76)90618-8
- GRIFFIN JE. Congenital Absence of the Vagina. Ann Intern Med. 1976;85(2):224. doi:10.7326/0003-4819-85-2-224
- Bjørsum-Meyer T, Herlin M, Qvist N, Petersen MB. Vertebral defect, anal atresia, cardiac defect, tracheoesophageal fistula/esophageal atresia, renal defect, and limb defect association with Mayer-Rokitansky-Küster-Hauser syndrome in co-occurrence: two case reports and a review of the literature. J Med Case Rep. 2016;10(1):374. doi:10.1186/s13256-016-1127-9
- Herlin M, Hojland AT, Petersen MB. Familial occurrence of Mayer–Rokitansky–Küster–Hauser syndrome: A case report and review of the literature. Am J Med Genet A. 2014;164(9):2276-2286. doi:10.1002/ajmg.a.36652
- Biason-Lauber A, Konrad D, Navratil F, Schoenle EJ. A WNT4 Mutation Associated with Müllerian-Duct Regression and Virilization in a 46,XX Woman. New England Journal of Medicine. 2004;351(8):792-798. doi:10.1056/NEJMoa040533
- Wottgen M, Brucker S, Renner SP, et al. Higher incidence of linked malformations in siblings of Mayer-Rokitansky-Kuster-Hauser-syndrome patients. Human Reproduction. 2008;23(5):1226-1231. doi:10.1093/humrep/den059
- Jacquinet A, Millar D, Lehman A. Etiologies of uterine malformations. Am J Med Genet A. 2016;170(8):2141-2172. doi:10.1002/ajmg.a.37775
- Opitz JM. Vaginal atresia (von Mayer‐Rokitansky‐Küster or MRK anomaly) in hereditary renal adysplasia (HRA). Am J Med Genet. 1987;26(4):873-876. doi:10.1002/ajmg.1320260414
- Shokeir MH. Aplasia of the Müllerian system: evidence for probable sex-limited autosomal dominant inheritance. Birth Defects Orig Artic . Published online 1978:147-165.
- Williams LS, Demir Eksi D, Shen Y, et al. Genetic analysis of Mayer-Rokitansky-Kuster-Hauser syndrome in a large cohort of families. Fertil Steril. 2017;108(1):145-151.e2. doi:10.1016/j.fertnstert.2017.05.017
- Azoury RS, Jones HW. Cytogenetic findings in patients with congenital absence of the vagina. Am J Obstet Gynecol. 1966;94(2):178-180. doi:10.1016/0002-9378(66)90460-1
- FRASER IS, BAIRD DT, HOBSON BM, MICHIE EA, HUNTER W. Cyclical Ovarian Function in Women with Congenital Absence of the Uterus and Vagina. J Clin Endocrinol Metab. 1973;36(4):634-637. doi:10.1210/jcem-36-4-634
- Shane JM, Wilson EA, Schiff I, Naftolin F. A preliminary report on gonadotropin responsivity in the Rokitansky-Küster-Hauser syndrome (congenitally absent uterus). Am J Obstet Gynecol. 1977;127(3):326-327. doi:10.1016/0002-9378(77)90478-1
- Strübbe EH, Willemsen WN, Lemmens JA, Thijn CJ, Rolland R. Mayer-Rokitansky-Küster-Hauser syndrome: distinction between two forms based on excretory urographic, sonographic, and laparoscopic findings. American Journal of Roentgenology. 1993;160(2):331-334. doi:10.2214/ajr.160.2.8424345
- Howard LA, Mancuso AC, Ryan GL. Müllerian Aplasia with Severe Hematometra: A Case Report of Diagnosis and Management in a Low Resource Setting. J Pediatr Adolesc Gynecol. 2019;32(2):189-192. doi:10.1016/j.jpag.2018.11.006
- Rall K, Barresi G, Wallwiener D, Brucker SY, Staebler A. Uterine rudiments in patients with Mayer-Rokitansky-Küster-Hauser syndrome consist of typical uterine tissue types with predominantly basalis-like endometrium. Fertil Steril. 2013;99(5):1392-1399. doi:10.1016/j.fertnstert.2012.12.002
- Marsh CA, Will MA, Smorgick N, Quint EH, Hussain H, Smith YR. Uterine Remnants and Pelvic Pain in Females with Mayer-Rokitansky-Küster-Hauser Syndrome. J Pediatr Adolesc Gynecol. 2013;26(3):199-202. doi:10.1016/j.jpag.2012.11.014
- Konrad L, Dietze R, Kudipudi PK, Horné F, Meinhold-Heerlein I. Endometriosis in MRKH cases as a proof for the coelomic metaplasia hypothesis? Reproduction. 2019;158(2):R41-R47. doi:10.1530/REP-19-0106
- Duncan PA, Shapiro LR, Stangel JJ, Klein RM, Addonizio JC. The MURCS association: Müllerian duct aplasia, renal aplasia, and cervicothoracic somite dysplasia. J Pediatr. 1979;95(3):399-402. doi:10.1016/S0022-3476(79)80514-4
- Pittock ST, Babovic‐Vuksanovic D, Lteif A. Mayer–Rokitansky–Küster–Hauser anomaly and its associated malformations. Am J Med Genet A. 2005;135A(3):314-316. doi:10.1002/ajmg.a.30721
- Oppelt P, Renner SP, Kellermann A, et al. Clinical aspects of Mayer–Rokitansky–Kuester–Hauser syndrome: recommendations for clinical diagnosis and staging. Human Reproduction. 2006;21(3):792-797. doi:10.1093/humrep/dei381
- Strübbe EH, Lemmens JAM, Thijn CJP, Willemsen WNP, van Toor BSJ. Spinal abnormalities and the atypical form of the Mayer-Rokitansky-Küster-Hauser syndrome. Skeletal Radiol. 1992;21(7):459-462. doi:10.1007/BF00190992
- Preibsch H, Rall K, Wietek BM, et al. Clinical value of magnetic resonance imaging in patients with Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: diagnosis of associated malformations, uterine rudiments and intrauterine endometrium. Eur Radiol. 2014;24(7):1621-1627. doi:10.1007/s00330-014-3156-3
- ACOG Committee Opinion Number 274, July 2002. Nonsurgical diagnosis and management of vaginal agenesis. Obstetrics & Gynecology. 2002;100(1):213-216. doi:10.1016/S0029-7844(02)02158-0
- Ulrich U. Mayer-von Rokitansky-Kuster-Hauser syndrome in association with a hitherto undescribed variant of the Holt-Oram syndrome with an aorto-pulmonary window. Human Reproduction. 2004;19(5):1201-1203. doi:10.1093/humrep/deh198
- Cremers CWRJ, Strubbe EH, Willemsen WNP. Stapedial Ankylosis in the Mayer-Rokitansky-Kuster-Hauser Syndrome. Archives of Otolaryngology – Head and Neck Surgery. 1995;121(7):800-803. doi:10.1001/archotol.1995.01890070086018
- DeLancey JOL. Anatomie aspects of vaginal eversion after hysterectomy. Am J Obstet Gynecol. 1992;166(6):1717-1728. doi:10.1016/0002-9378(92)91562-O




